Every B cell receptor sequence in a repertoire came from a V(D)J recombination of germline genes. Each individual has only certain alleles of these genes in their germline, and knowing this set improves the accuracy of all aspects of BCR sequence analysis, from alignment to phylogenetic ancestral sequence reconstruction. This germline allele set can be estimated directly from BCR sequence data, and it’s time to treat such estimation as part of standard BCR sequence analysis pipelines.
This central message is not new, but it’s worth emphasizing because doing germline set inference is not part of most current studies of B cell receptor (BCR) sequences.
Indeed, the most common way to annotate sequences is to align them one by one to the full set of alleles present in the IMGT database, which has hundreds of alleles. Each individual has only a fraction of these alleles in their genome.
Unsurprisingly, aligning sequences one by one to the whole IMGT set can cause problems. Imagine that A and B are two germline alleles in IMGT that are similar to one another. Sequences deriving from germline allele A can somatically hypermutate to look more similar to the B allele than the A allele from which they came. If we allow A and B in our germline repertoire, such sequences will be incorrectly annotated as being from B when they are from A. This will certainly lead to an incorrect estimation of the naive sequence from which they came.
In addition, it’s known through the work of many groups that the total set of germline genes is much larger than that represented in IMGT. This is not surprising given that this region is tricky to sequence directly, and that so far genetic studies have been primarily done on people of European ancestry. Here again, if we are missing a sequence from our germline set, we will have problems with all of our downstream analyses.
Thus, we should be estimating per-sample germline sets for BCR sequence data. This is not a trivial task. In 2010, Scott Boyd and others were the first to use high-throughput sequencing data of rearranged BCRs to estimate per-sample germline sets with a combination of computation, expert judgement, and statistics. In 2015, the Kleinstein group made a big step by developing TIgGER, an automated method for inferring germline sets that weren’t too far from existing alleles, and more recently the Hedestam group developed IgDiscover, a method that could start more “from scratch” for species where we have little or no germline information.
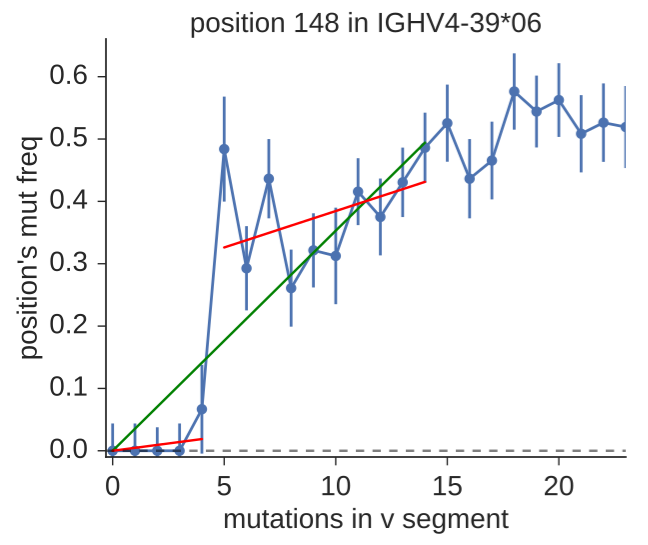
The motivation for Duncan’s work came from analyzing sequence data from diverse sources, and seeing clear evidence of alleles that were not represented in IMGT. He tried the existing tools but became frustrated first with software usability. He then started by re-implementing TIgGER, and then realized that he could use the same input information (their “mutation accumulation” plot depicted above) but in a way that more directly tests for the presence of new alleles, by considering the goodness of fit for one- vs two-component fits. In classic Duncan fashion, he has done a ton of validation, varying many different parameters in his simulation and also comparing the results of the different methods on experimental data sets. The work is now up on arXiv and is part of his partis suite of repertoire analysis tools.
There’s still a lot to be done here, and our knowledge of this highly diverse and important locus will continue to improve as more sequencing data of all types comes in. This is one example of many showing how analysis of a whole data set at once is more powerful for each individual sequence than one-at-a-time analysis of sequences.