High-throughput sequencing of our adaptive immune repertoires holds great promise for understanding immune state. These sequences implicitly contain a wealth of information on past and present exposures to infectious and autoimmune diseases, to environmental stimuli, and even to tumor-derived antigens. In principle, we should be able to use these sequences of rearranged receptors to infer their eliciting antigens, either individually or collectively.
We’re starting to see neat progress in these areas for T cell receptors (TCRs). Some recent studies compare TCR repertoire between individuals who do or do not have some immune state, such as an immunization, an autoimmune disease or a viral infection and work to find sequence-level differences between the repertoires. The Walczak-Mora team recently upped the bar by not requiring a control cohort. There has also been interesting progress on predicting epitope specificity from TCR sequence using structurally-informed sequence analysis.
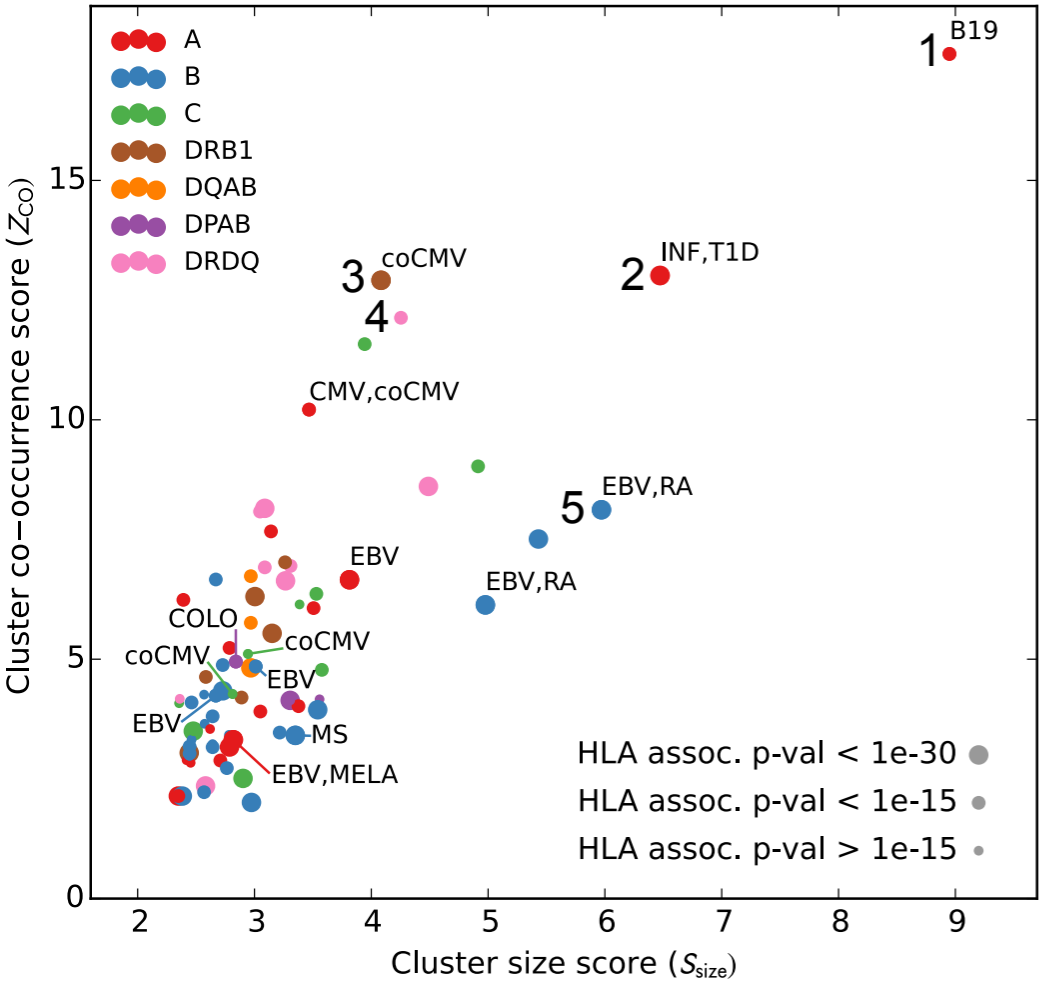
Phil Bradley, just down the hall from us, wanted to take a different approach, asking given appropriate statistical analysis of a sufficiently large data set, can we infer pathogen-responsive TCRs from co-occurrence and HLA information alone? (If you don’t remember about HLA, it determines the sequence of MHC, the hot dog bun presenting peptides for recognition to T cells.) He showed that this indeed was the case, one example of which is shown in the figure above. Each point is a cluster of TCR sequences, where clustering is performed based on both co-occurrence and on TCR sequence similarity. Only TCR sequences that are significantly associated with an HLA type are allowed to participate in the clustering, and only clusters that were significant in terms of family-wise error rate are shown. These clusters are plotted with respect to the cluster size and a co-occurrence score.
{kind=link}
The surprising result is that this procedure, which knows nothing about what stimulated the TCRs to expand, identifies previously-labeled TCR sequences corresponding to certain immune states. You probably recognize EBV, MS, and CMV, but we also see B19=parvovirus B19, INF=influenza, RA=rheumatoid arthritis, T1D=type 1 diabetes, and others. That’s pretty neat! This, along with other fun surprises, is published in eLife.
I made very minor contributions to this manuscript, but wanted to write about it because I think it’s an exciting advance. This proof of concept is definitely motivating us to think harder about what sorts of statistical frameworks would be useful for doing this sort of research more comprehensively. Thanks to Will and Phil, to the Hansen lab for the neat data, and to the study participants.