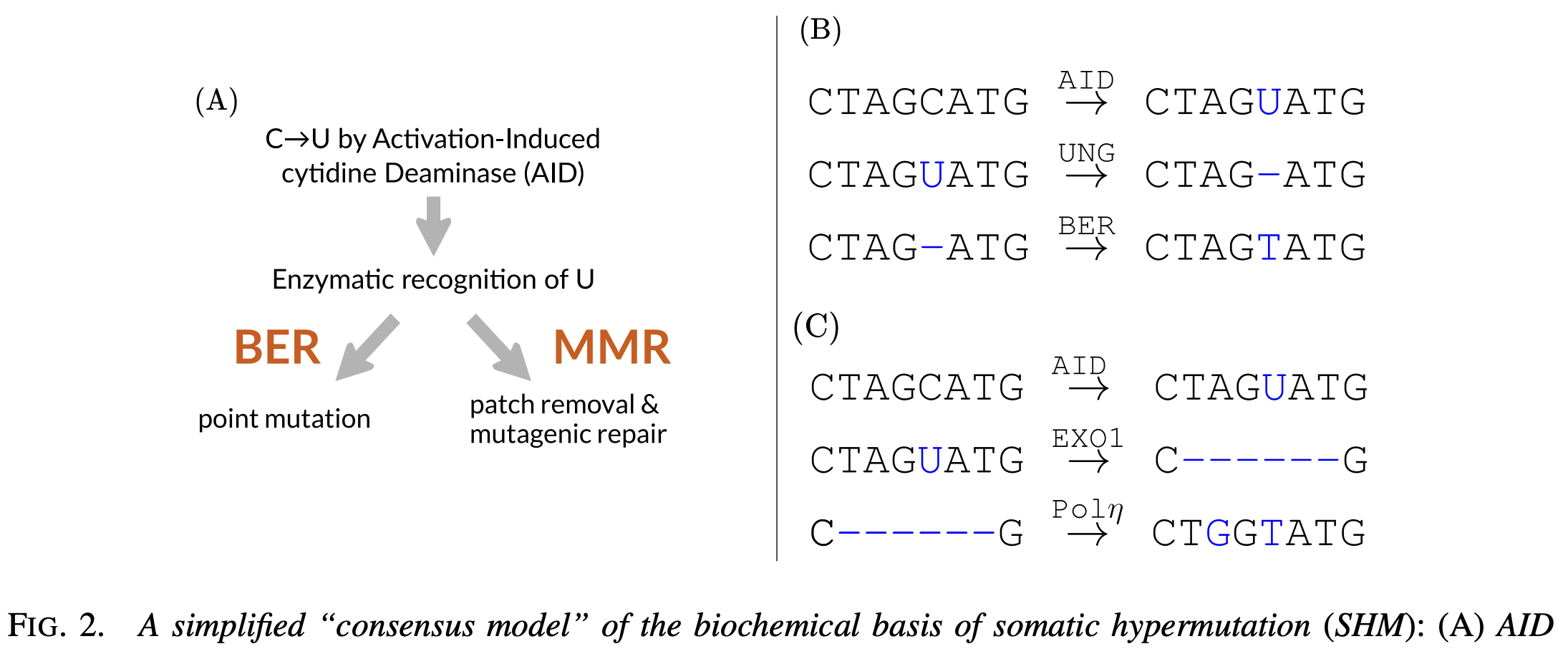
Somatic hypermutation (SHM) is one of the craziest things I’ve ever heard of: we have enzymes that intentionally damage and mutate our DNA! This process is the foundation of the affinity maturation process that enables us to generate high-affinity antibodies against just about any target. Without it, we’d be limited to “just” random recombinants of germline genes.
Our SHM work was motivated by a desire to reconcile statistical analysis of somatic hypermutation with what we know mechanistically from lab experiments. Whenever one can bring reality into modeling there is the potential for a double win: one can learn something about underlying mechanism, and one can get better inferences by using a model that reflects underlying reality.
My interests were piqued by several fascinating papers. On the computational side there were Spisak et al 2020 and Zhou and Kleinstein 2020 who found evidence for an effect of absolute position along the sequence in SHM rates. The Spisak paper in particular showed a very irregular pattern of per-site effects that seemed surprising. One the lab side there was Wang 2023, which showed lab evidence for “mesoscale” effects in which sequence 50-ish bases away from a focal site can influence mutation at that focal site.
The Spisak paper also quantified substantial spatial co-localization of mutations: mutations are more likely to appear in clusters than under a model of independent mutation of sequences. This part is easier to explain by one of two mechanisms. One mechanism is that AID, the initiator of DNA damage, can slide along and make groups of mutations. The second mechanism is that this same enzyme can damage DNA stimulating a cascade of downstream repair events.
The Spisak paper also built a nice framework for further computational study of SHM: a phylogenetic one! They found clonal families in marvelous big datasets that were entirely unproductive. This meant that the VDJ recombination process led to an out-of-frame rearrangement that was carried alongside a productive rearrangement, mutating but not being expressed or selected.
Thus the time felt right. We had some big new datasets, a backpack full of great stats/ML tools, and we were ready to unite the mechanistic and statistical perspectives on SHM.
Our first effort was all-in: parameterize a mechanistically-explicit model of somatic hypermutation, and fit it using data. By such a model I mean one that has a probability of AID damage and procession, and of the various error-prone repair pathways. If we had such a perfect model in hand, it should be more predictive than a local-sequence-context model. The model would be an interesting result in itself, attaching probabilities and parameters to the current consensus model of SHM.
To say this project was hard would be substantial understatement. One way statistical inference becomes difficult is when there are lots of possible ways the observed data can come to be. To calculate a likelihood under such a model one has to integrate over all those possible ways. In our case, we could have an AID event here, and then a nucleotide stripping event all the way over to there, or the AID event could have happend somewhere else (and perhaps faithfully repaired), and then… you get the idea.
But, never fear, neural networks and simulation-based inference are here! We tried many formulations, first starting with a CNN working directly on one-hot encoded sequence, then an approach in which we tried to infer a latent state describing the events leading to the observed sequence. The signal was really too weak, and the data too noisy, for these approaches to work.
What ended up working was to hand-design some summary statistics about the sequence (e.g., how close pairs of mutations are to one another) and then use those as inputs to a neural network that predicted the parameters of our model. This model is trained using simulation.
The reward for this project was the parameter estimates themselves. We found less evidence of processivity than is measured in vitro, a slight bias towards the BER pathway over the MMR pathway, and a slight strand bias. However, this project did not deliver the “double win” I was hoping for, as simpler context-based models are quite superior to our mechanistic models for prediction.
Many kudos to Thayer Fisher for overcoming many hurdles for this sometimes-arduous project, which lasted most of his PhD, and to Noah Simon and Julia Fukuyama for guiding us through the statistical jungles. The paper is now published in Annals of Applied Statistics and you can find the PDF here.
As it was becoming clear that the mechanistic project wasn’t going to deliver more accurate mutability predictions than the state of the art, we started thinking about alternative paths. Still hoping for a semi-mechanistic model, we worked fairly hard on a project in which we applied penalized regression to identify mutation hotspots that would influence nearby bases. This did not work out. The core problem was that we needed to do something to handle the problem with wide-context models: if we give a parameter to every k-mer, we end up with an exponential number of parameters.
We ended up solving this problem with a “tiny convolutional neural network” approach. The idea is that 3-mers are quite informative for SHM rate estimation, and there aren’t that many of them. However, we want to use a wide context. The solution is to train an embedding of the 3-mers which then gets consumed by the convolution layer and turned into prediction.
The upshot? A linear rather than exponential scaling in the parameter count with the size of the window. So we get to have 13-base-wide models for the parameter cost of a 5-mer mutation model.
But I want to make it clear that the improvement that we get over a simple 5-mer model, like the S5F model, is small. Did we try more complex alternatives? Why yes we did! We tried per-site rates, transformers, positional embeddings, etc. Nothing helped above the base model.
In the end this may tell us something about the biology, which was one of my original goals: if there are effects of per-site rates or mesoscale-level contexts, they are well-enough modeled by more local sequence context that they don’t measurably improve prediction.
However, this conclusion is tempered by the fact that this problem appears quite data-limited. We found evidence that there was a reasonable amount of error in the marvelous big data set, and despite heroic efforts by Mackenzie (documented in the paper) to find additional data, we only had a few reasonably sets with a substantial number of out-of-frame rearrangements. [If you are reading this and are doing a bunch of genomic DNA sequencing, rather than RNA sequencing, and have substantial data sets, please get in touch!] I hope that we get to revisit this problem with more data and train richer models.
This paper is now available at eLife.
These projects have laid the foundation for our current adventure, which is to fit models of natural selection on antibodies. That clearly merits complex models (antibody structure and function is complicated!) and we have all the productive data in the world to fit them.
For all of these projects, I want to very much thank co-authors Kevin Sung, Mackenzie Johnson, Will Dumm, and Hugh Haddox in addition to those mentioned above. We are tremendously grateful to funding for this project from the NIH and HHMI.
You made it to the end. Congratulations! ☕️
Comments? Head on over to b-t.cr.