I get a little bubble of excitement every time I encounter someone who doesn’t know about antibody affinity maturation. When I do, I get to point at my neck lymph nodes and describe that there is this little Darwinian process inside you that evolves antibodies to have higher affinity (i.e. bind better) against whatever just made you sick. Isn't that amazing? I will accept public speaking slots or alumni panels just so I can tell more people about it.
But the reality of affinity maturation is more interesting and complicated than this cursory description would suggest. It’s a noisy process of mutation and selection, and if you aren’t careful, it can actually be difficult to discern the signal of natural selection from the mutation biases! That may come as a surprise for people who only hear the short version.
Let’s juxtapose three recent manuscripts from our group and discuss this phenomenon.
In the replay project, we had a special opportunity to understand selection in a system we could observe in great detail. In a Victora lab tour de force, Tatsuya and Ashni were able to sequence 119 replicate germinal centers, all starting from the same naive B cell receptor and responding to the same antigen. We could quantify the amount by which mutations improve binding against the target antigen using a yeast display experiment by Tyler Starr. Ashni conducted an experiment with a passenger allele that gave us a custom probabilistic model of somatic hypermutation specifically for our naive B cell receptor. (This is crazy.)
Here’s the result from Figure 3 of that preprint. On the x axis is affinity change of a given mutation, and on the y axis is how frequently we saw it. The correlation is… underwhelming. If we had such strong affinity selection in the germinal center, shouldn’t this look stronger?
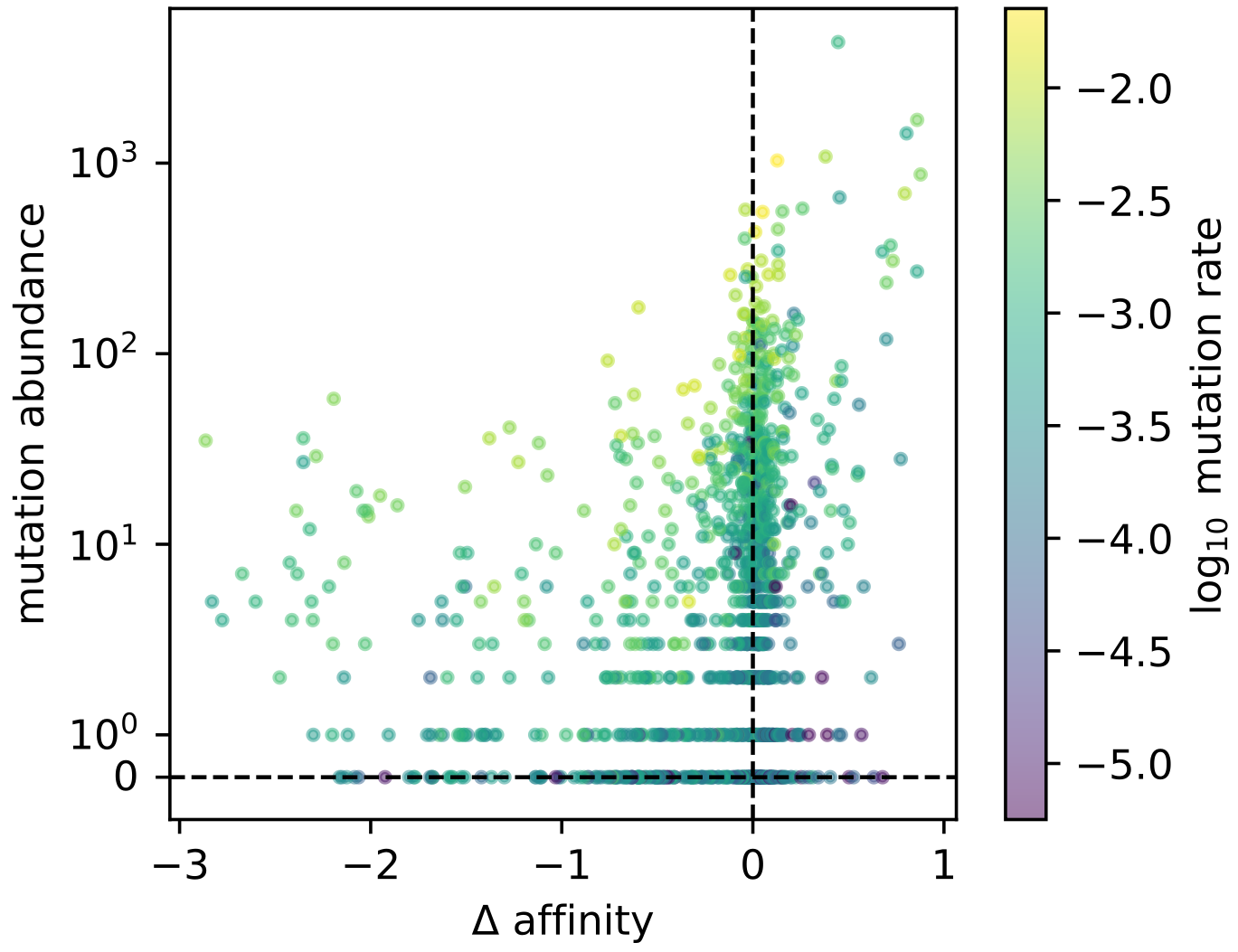
On the other hand, things look a lot nicer when we plot mutation rate on the x axis.
We can see the effect of the affinity changes (now colors).
Selection pushes the blue (positively selected) points above the y=x line, and it pushes the red (negatively selected) points below the y=x line.
Nice!
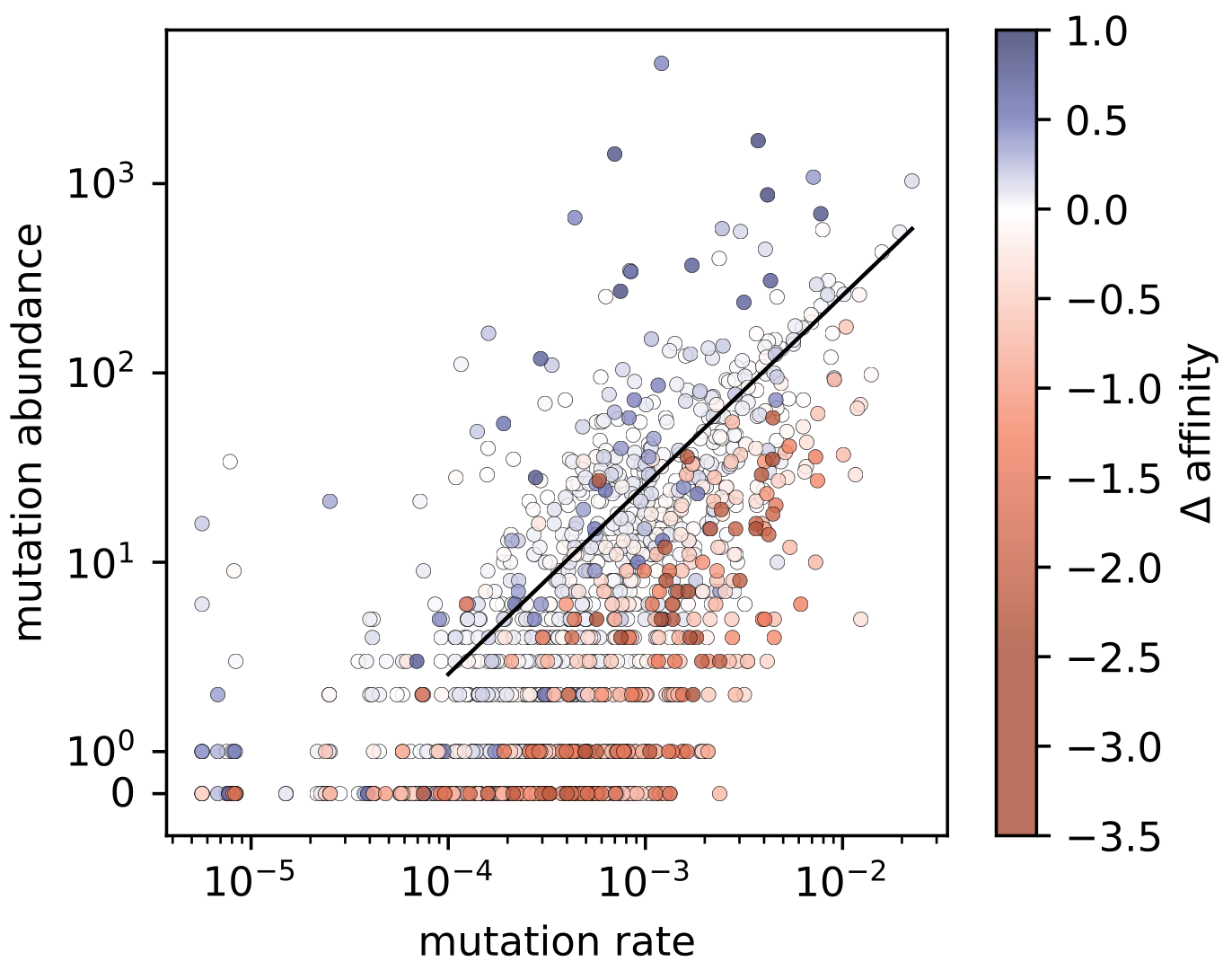
In summary, selection matters, certainly. But we can only see its effect once we take somatic hypermutation rates into account!
We were led to another perspective on mutation versus selection from a different source: protein language models. Like many, we were impressed by the results of Hie et al 2024 who showed that in their system, they could achieve increased binding affinity through directed evolution guided by a general protein language model. If that’s the case, then surely we should be able to see solid signal from that model in the evolutionary process of natural affinity maturation.
With that motivation, in our new Johnson, Sung, et al. 2025 preprint, we built a framework called EPAM (Evaluating Predictions of Affinity Maturation) to compare different approaches for predicting antibody evolution. EPAM is a unified framework that can take a diverse collection of models and standardize their predictions for direct comparison. We evaluated models across three categories: pure SHM models like S5F, combined SHM+selection models using protein language model selection factors, and pure protein language models like AbLang2 and ESM-1v. These were then tested on large human BCR repertoire datasets as well as the controlled mouse replay experiment.
SHM dominates! The classic S5F model, which uses a 5-nucleotide context to predict mutation probabilities, substantially outperformed both AbLang2 and ESM-1v in predicting where amino acid substitutions would occur. Our newer “Thrifty” SHM model, with its wider 13-nucleotide context, performed even better—achieving 0.834 versus 0.784 overlap scores compared to S5F, while using orders of magnitude fewer parameters than the protein language models.
This raised an obvious question: if nucleotides are so important, why don’t we train a nucleotide model on productive sequences? Well, it turns out that a Thrifty-like model with a few thousand parameters significantly outperformed state-of-the-art protein language models with millions of parameters, giving us the best model in this benchmark. Furthermore, when we tried to improve the SHM models by adding selection factors from sophisticated protein models like ESM-1v, performance actually got worse.
Returning to the replay experiment described above, we showed that even in this highly controlled setting with tailored models for both SHM (ReplaySHM) and selection (deep mutational scanning), the selection model provided only modest improvements over the SHM model. Here again, even when we have an experiment providing exactly the quantities needed to infer natural selection, SHM dominates the process.
But we don’t have to choose between mutation or selection…
Computational evolutionary biologists have been separating mutation from selection for decades. This selection inference is generally called “dN/dS” analysis, and it’s standard enough to appear in Wikipedia. We ourselves have applied this sort of approach to B cells. However, this previous work has a shortcoming: it operates on the level of sequence alignments (classic dN/dS analysis), or alternatively pairwise alignments with known germline antibody genes (our B cell paper). What if we want to make predictions on full individual antibody sequences like language models do? Could we give this classical framework a transformer-based upgrade?
We can.
Our new model is called a Deep Natural Selection Model (or DNSM) and has just come out in MBE. Unlike classical selection models, we can make sitewise predictions on single antibody sequences. Unlike protein language models, the DNSM only quantifies natural selection rather than a conflation of mutation and selection.
What does this model teach us? First, the pattern of natural selection on antibodies is complex! It is certainly not just a question of CDR versus framework regions. The patterns vary a lot from site to site and between sequences.
This model is able to tell us things that previous models cannot. Because we can estimate selection on individual sequences, we can look at how patterns of natural selection change as the sequence changes. And they do (Figure 6 in the paper)!
Furthermore, because we don’t need to align sequences, we can estimate natural selection on sites in the varying-length CDR3. One of my favorite things we learned in this project is the consistently purifying selection on the aspartic acid which frequently appears just before the end of the CDR3 (Figure 7 and S9 in the paper). Did I mention that natural selection for antibodies is complex?
I forgot to put an image in the paper that shows the structural reason for this, so here it is:
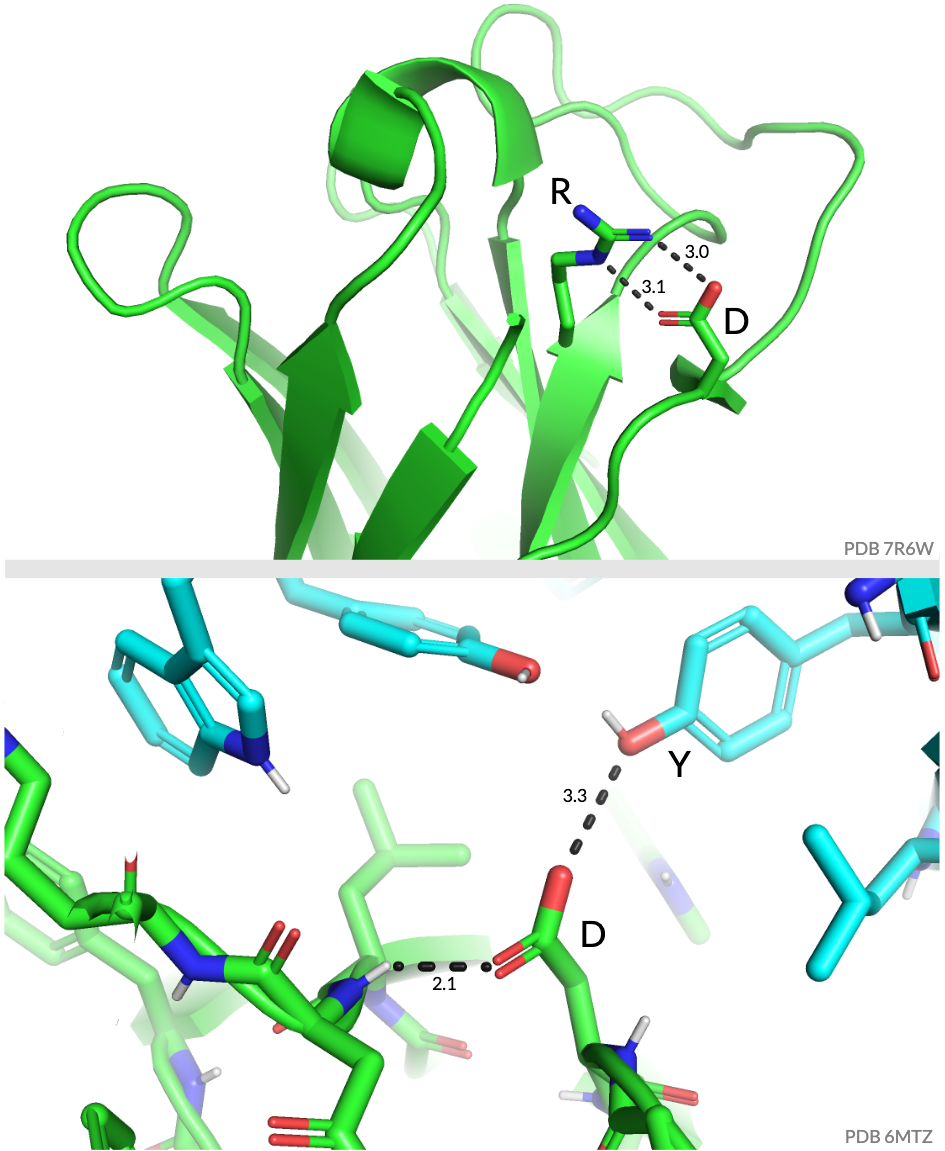
The upper panel shows aspartic acid (D) forming a salt bridge across the CDR3. The lower panel shows another setup in which the D is interacting with the backbone of the CDR3 and also with a tyrosine (Y) extending to the light chain! These presumably are important for stabilizing the antibody, and so are under strong purifying selection. (Phil, Tyler and Dave found these structures, and Hugh made the figure.)
I want to make it clear that this DNSM model is not yet morally equivalent to a language model. Specifically, it only makes a per-site prediction of fitness, whereas language models make a per-site prediction for each alternate amino acid. We have adapted the model to be such a mutation-selection model, and I have just submitted the corresponding paper. You can get a preview, though, on this AIRR Community Seminar.
Why does selection feel so weak relative to mutation in affinity maturation?
Let’s look at Figure 2B from the replay paper. This figure shows all possible amino acid replacements to the naive BCR sequence used in that paper (not just those that appeared in the sequenced mice), with each mutation represented as a dot in the scatter plot. On the x axis is how the replacement changes affinity, and on the y axis is how it changes expression (the ability of the cell to make the antibody).
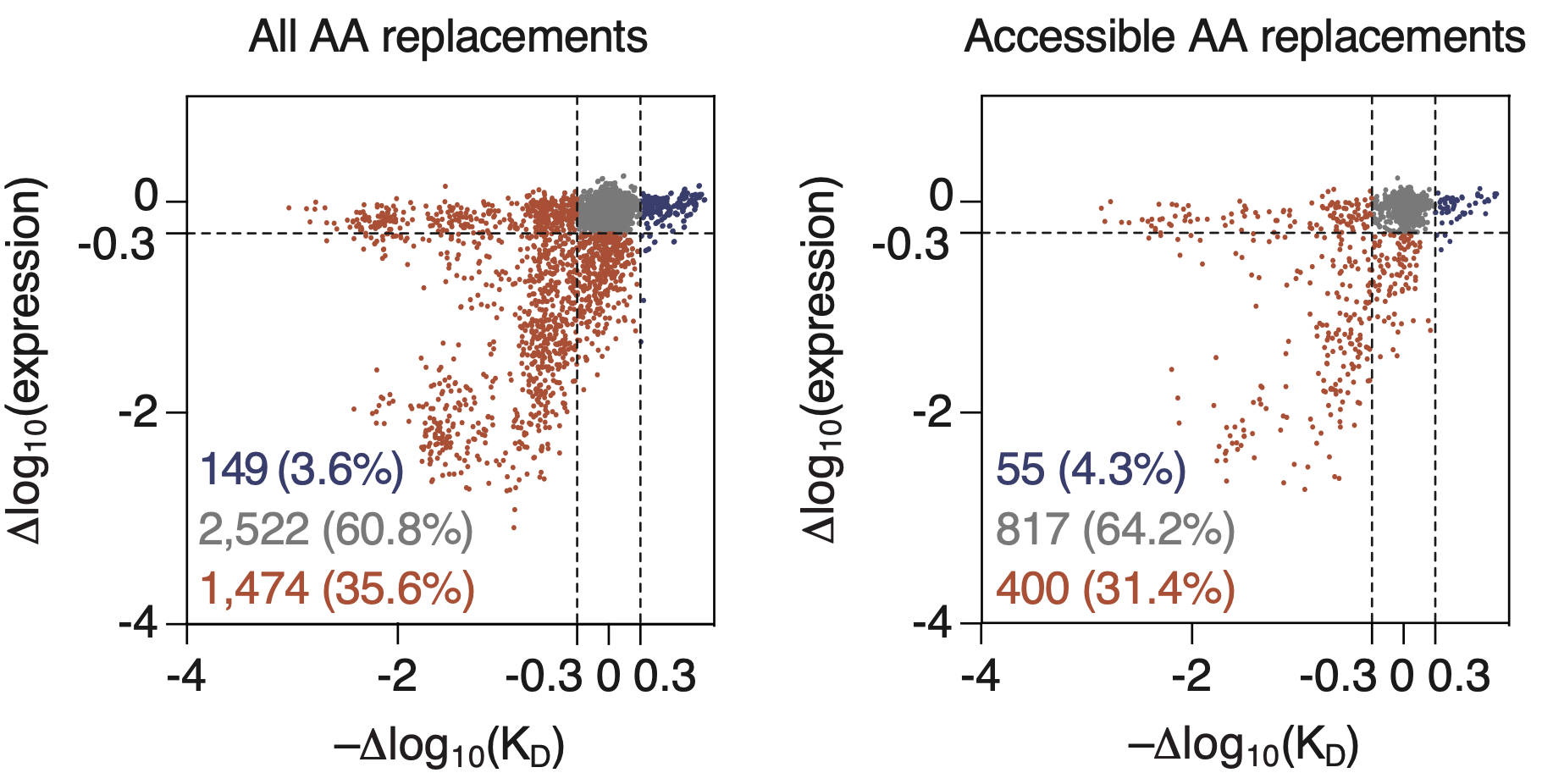
Those blue points are the replacements that will advance affinity maturation: the ones that are better binders and don’t lose too much expression. As shown, that’s a measly 3.6% for all replacements and 4.3% for the amino acids that are accessible via a single nucleotide change!
For an evolving B cell to survive its perilous journey and sample several beneficial mutations that will increase its affinity, it must avoid sampling those detrimental red points. Along the way it’s going to sample a ton of gray mutations, which are approximately neutral. This is why we see so many mutations that do not improve fitness. Furthermore, mutation patterns are highly nonuniform, meaning that the mutation process feels more powerful in these many neutral mutations. Because language models are trained on collections of observed sequences, they get quite distracted by these mutation patterns.
This was a blog post talking about our work, not a research paper.
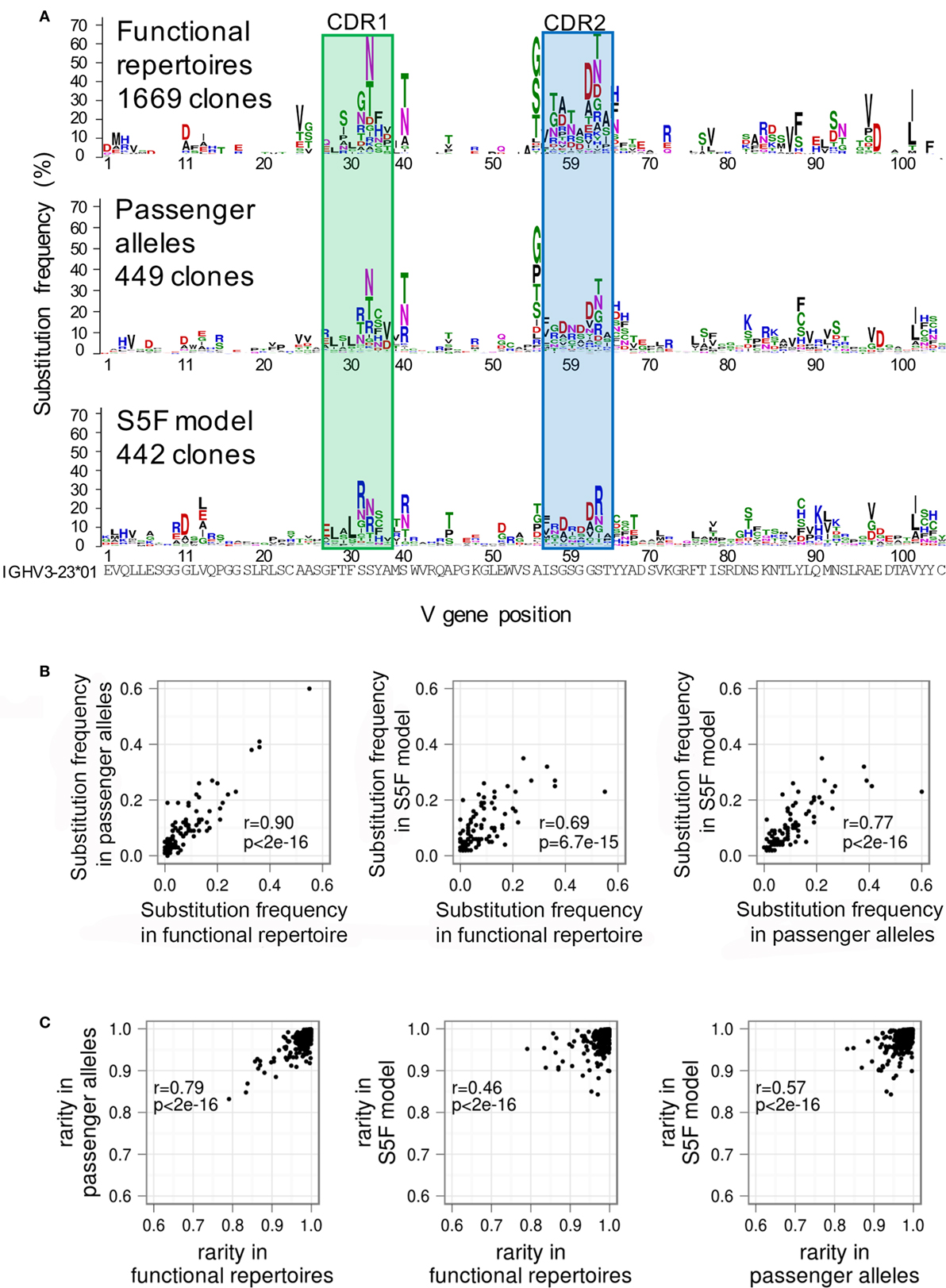
I have not discussed the many antecedents to our work, which I hope we do a good job of citing in our papers. Nevertheless, I would be remiss if I did not mention how the Kleinstein lab and the Mora-Walczak group have done a lot of important related work. The Sheng-Schramm GSSP paper figure 5 has been a consistent inspiration: just look at that agreement between the functional repertoires and the neutrally evolving passenger clones!
{kind=link}
Also, it turns out that van Ginneken 2025 independently developed some of the same ideas as our EPAM project after reading the Hie paper.
Thank you to everyone who worked on these projects! I had a blast.
Special appreciation to our funding from the NIH and HHMI.
Comments? Head on over to b-t.cr.