Can we predict how sites of an antibody will tolerate amino acid substitutions? Kristian Davidsen posed this question shortly after he arrived in my group, pointing out that being able to do such prediction would be quite useful. For example, engineered antibodies sometimes aggregate into clumps or have other properties that that make them useless for mass production. If we could figure out ways to change the amino acid sequence of an antibody without changing binding properties, that could help us avoid aggregation and make a more useful antibody.
How to start to address this complex and high-dimensional question? Although people have started to do deep mutational scanning on antibodies this type of data is hard to come by. On the other hand, B cell repertoire (i.e. antibody-coding) sequence data is becoming plentiful. B cells undergo affinity maturation to improve binding in collections of sequences called “clonal families” grouped by naive ancestor sequence (more background here). Although it’s not quite the same, we can use the frequency of an amino acid at a given site in that clonal family as a proxy for the suitability of that amino acid for an antibody binding the same target. Or perhaps such a clonal-family amino-acid frequency is simply an interesting object in itself.
In any case, our goal became: given a single sequence from a clonal family, can we predict the amino acid frequency of the collection of sequences in the clonal family? We follow Sheng, Schramm et. al (2017) in calling this sort of thing a substitution profile. Inferring a substitution profile from a single sequence might sound hard or impossible, but several features of the affinity maturation process lean in our favor:
- There are a finite number of germline ancestor sequences from which diversification begins, and we can do a good job of inferring from which ancestor a given B cell sequence derives.
- Simply because of the mutation process, some sites are more likely to mutate than others (recently covered here).
- There’s lots of other repertoire data that we can use to watch the affinity maturation process.
This last one is sort of special, and deserves a bit of explanation. If we had a database containing every B cell sequence that had ever occurred, one could simply look for clonal families containing the sequence given to us, and take the average amino acid profile of those clonal family sequences. Unfortunately we don’t have access to such a database, but we can at least look for somewhat similar sequences and learn from their substitution profiles.
The previous Sheng-Schramm work, as well as contemporaneous work by Kirik et. al (2017), also indicates that various germline genes diversify in various characteristic ways (this sentiment also appears in Duncan’s first B cell paper and I’m sure many other previous works). This tells us that a profile based on germline gene identity should also inform a predicted substitution profile. Also, the context-sensitive neutral process given a germline gene should be helpful.
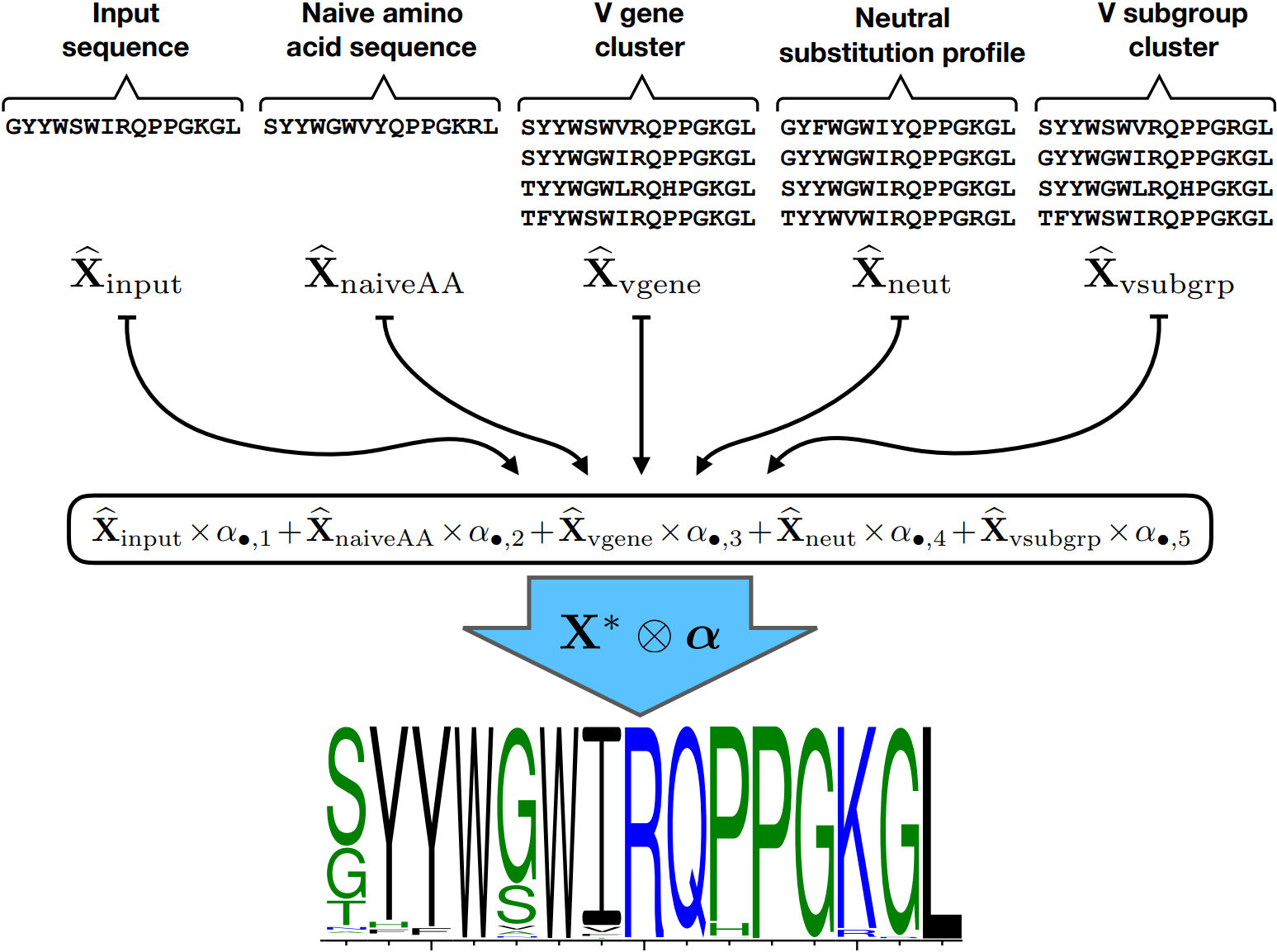
How do we combine these various sorts of information, especially considering that what is helpful for prediction at one site might not be helpful for another? Well, our group, consisting of Kristian, Amrit Dhar, and Vladimir Minin, decided to use a penalized tensor regression framework. That sounds fancy, but it just means that a single profile is a weighted linear combination of the profiles from each of the sources of information (see picture above). The weights may differ from site to site, but the kind of penalization we put on keeps them from changing too much between neighboring sites. It also zeroes out coefficients that don’t seem to be helping out-of-sample prediction. We find that different sources of information are useful for different parts of the B cell receptor sequence, in a way that corresponds to intuition about the “framework” and “complementarity determining” regions.
In any case, we show that integrating these diverse sources of information can help prediction, and provide a pre-trained prediction algorithm to do so. The code and parameters are on Github and the paper is on arXiv. So have at it with your sequences, and let us know how it fares!
I think that predicting substitution profiles is an interesting and useful goal. It did take a little getting used to, because we previously worked super hard to get per-residue natural selection estimates for B cell receptors by carefully separating the mutation and selection processes; here these substitution profiles just smash all that complexity down to a simpler object. There’s more to be done here: as data sets get bigger and machine learning algorithms get smarter, I look forward to seeing prediction improve! Thanks to Amrit, Kristian, and Vladimir for a fun project.